Batch effects are generated by sequencing depth, but not sequencing replicates alone
Benjamin R Babcock
Sequencing_Effect.RmdIdentical (duplicate) Sequencing
Processing the Seurat Object
Data filtering & normalization
library(BatchNorm)
# Import unfiltered Seurat object (included with 'BatchNorm' package)
data(package = 'BatchNorm', PBMC4)
# Run "standard" Seurat workflow:
# Including filtering by mitochondrial percentage (+5 SD)
# Including data normalization, variable gene selection and gene scaling (performed on all samples together)
PBMC4 <- PBMC4 %>%
MitoFilter() %>%
NormalizeData(normalization.method = "LogNormalize", assay = "RNA", scale.factor = 10000) %>%
NormalizeData(verbose = FALSE, assay = "ADT", normalization.method = "CLR") %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData() %>%
RunPCA(npcs = 30)Selecting the appropriate number of Principal Components for UMAP reduction
## Identify correct numbers of PCs
## (Takes up to 5 minutes. Not run while rendering vignette for time)
#PBMC4.pca.test <- TestPCA(PBMC4)
#PBMC4.pca.test[, 1:20]
## 16 PCs with z > 1
## Proceed with 16 PCs for dimensional reduction & clustering
## Visualize PCs plotted by standard deviation:
ElbowPlot(PBMC4)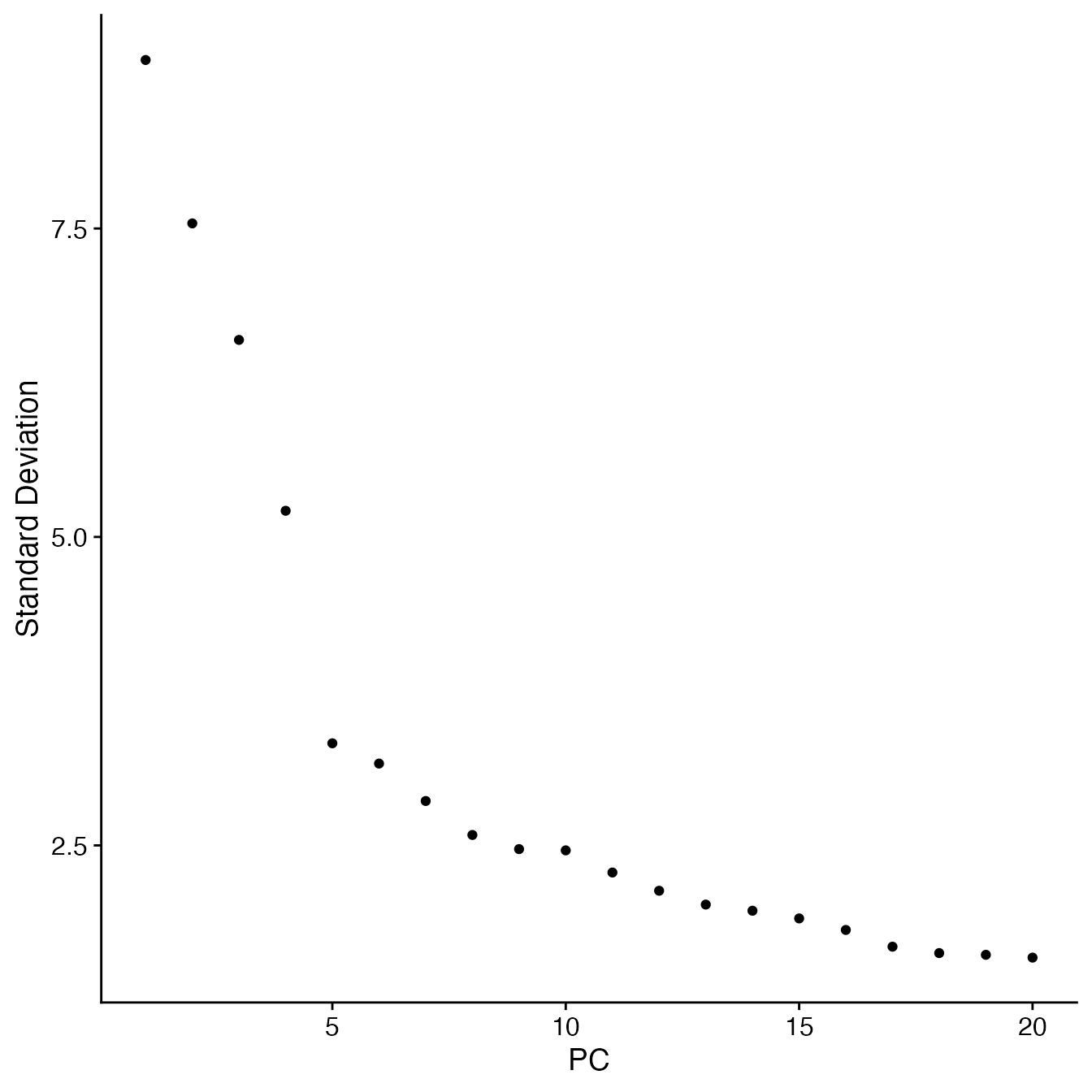
Generating a UMAP and clusters
PBMC4 <- PBMC4 %>%
RunUMAP(reduction = "pca", dims = 1:16) %>%
FindNeighbors(reduction = "pca", dims = 1:16) %>%
FindClusters(resolution = .8)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 12721
## Number of edges: 437773
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8794
## Number of communities: 21
## Elapsed time: 1 seconds
UMAPPlot(PBMC4, cols = colors.use, group.by = "orig.ident") + ggtitle("Default Workflow")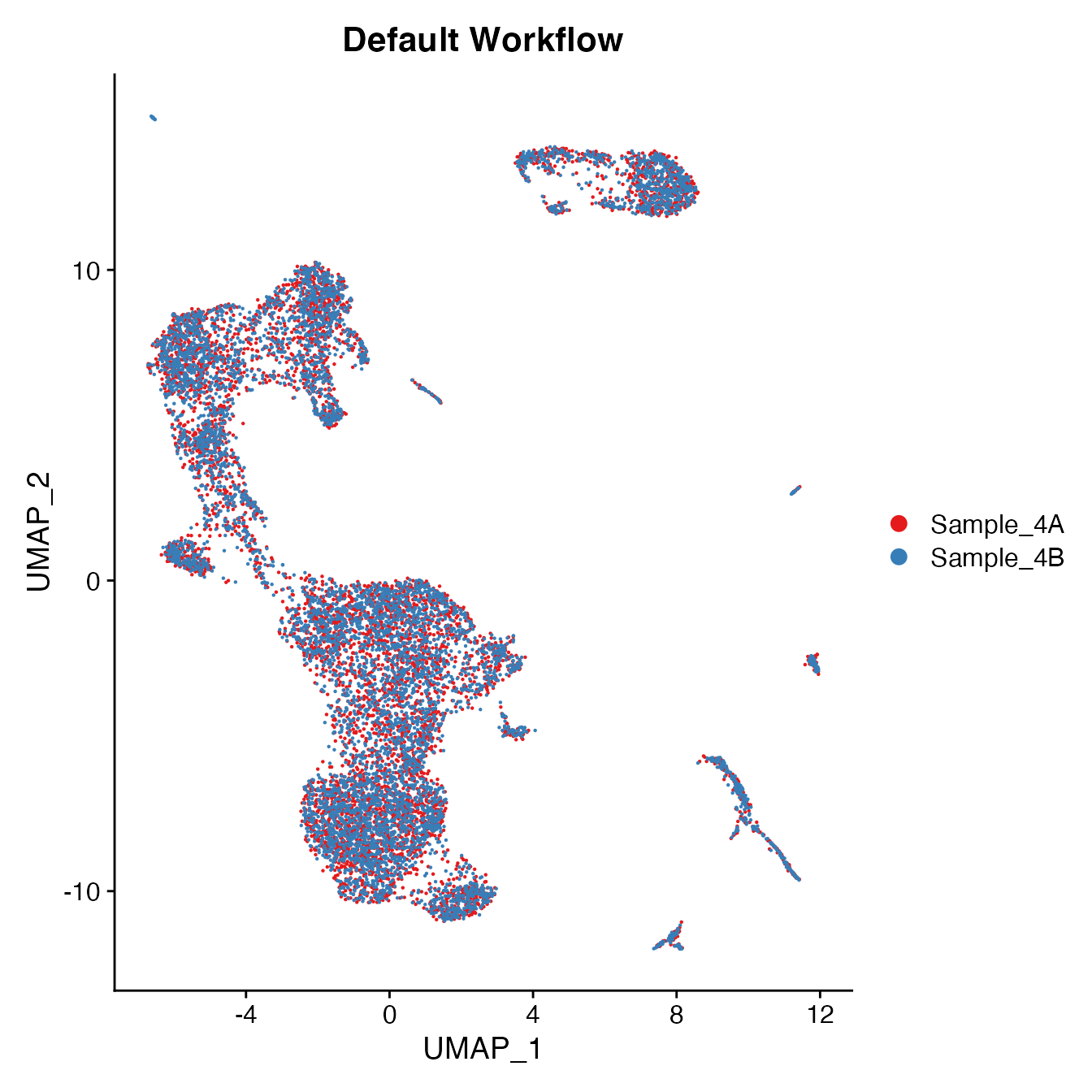
Measuring sample-UMAP integration (generating an iLISI score)
GetiLISI(object = PBMC4, nSamples = 2)## [1] 0.03041182Cell Typing of joint PBMCs object for CMS
Convert cluster classifications to cell type classifications
# For complete cell classification workflow see our vignette "Biaxial Gating of a Single Sample"
# More details can be found in figure S3 of our manuscript "Data Matrix Normalization and Merging Strategies Minimize Batch-specific Systemic Variation in scRNA-Seq Data."
UMAPPlot(PBMC4, cols = colors.use, pt.size = 2,
group.by = "seurat_clusters", label = T)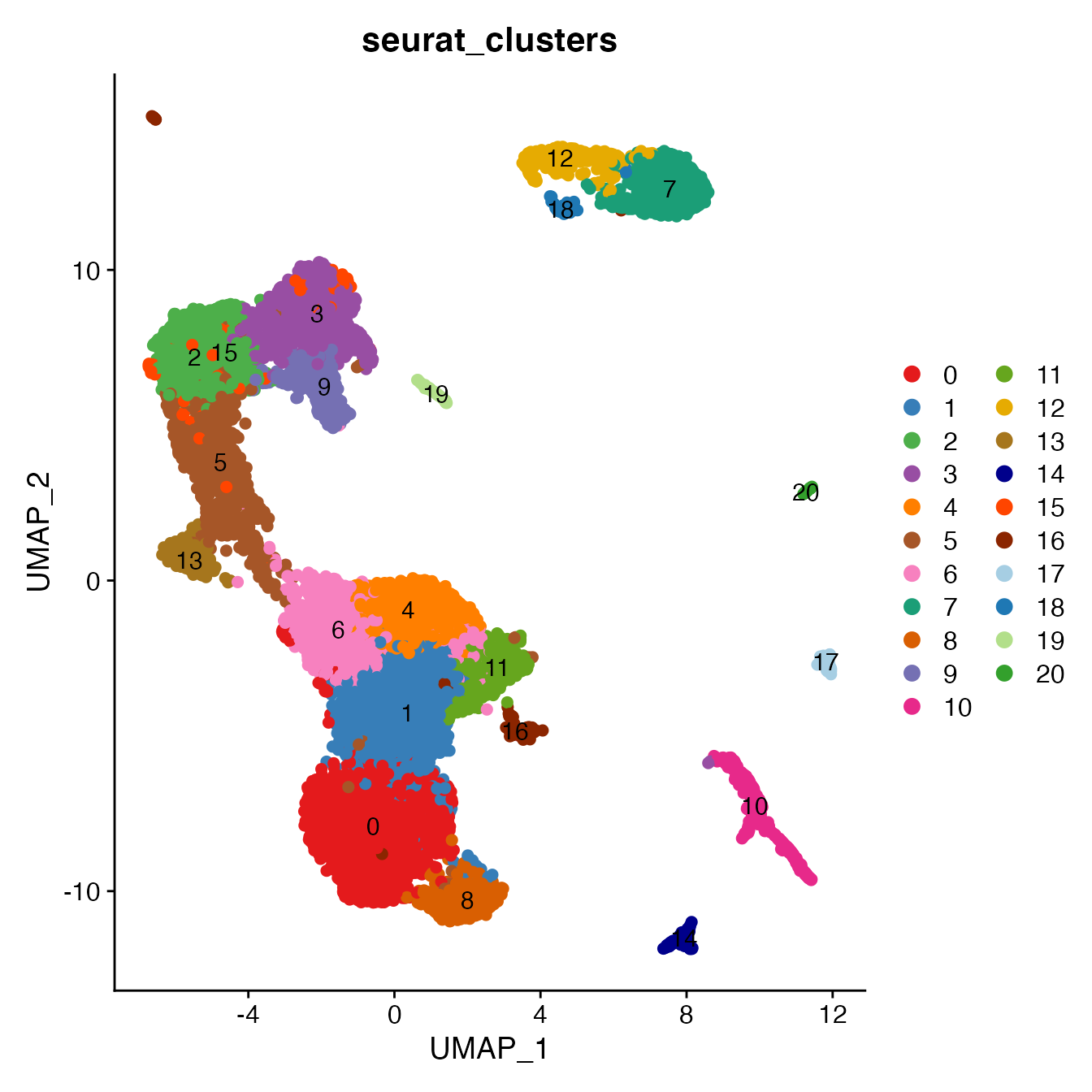
# B_Naive = 7
# B_Memory = 12, 18
# T_CD4 = 0, 1, 4, 6, 16
# TReg = 11
# T_CD8 = 8, 13
# NK_T = 5, 15
# NK = 2, 3
# NK_CD56Hi = 9
# Monocyte_Classical = 10
# Monocyte_NonClassical = 14
# Dendritic_Cells = 17
# HSPCs = 20
# Cycling_Cells = 19
Idents(PBMC4) <- PBMC4[["seurat_clusters"]]
Idents(PBMC4) <- plyr::mapvalues(Idents(PBMC4), from = c(7, 12, 18, 0, 1, 4, 6, 16,
11, 8, 13, 5, 15, 2, 3, 9,
10, 14, 17,
20, 19),
to = c('B_Naive', 'B_Memory', 'B_Memory', 'T_CD4', 'T_CD4', 'T_CD4', 'T_CD4', 'T_CD4',
'TReg', 'T_CD8', 'T_CD8', 'NK_T', 'NK_T', 'NK', 'NK', 'NK_CD56Hi',
'Monocyte_Classical', 'Monocyte_NonClassical', 'Dendritic_Cells',
'HSPCs', 'Cycling_Cells'))
Idents(PBMC4) <- factor(Idents(PBMC4),
levels = c("B_Naive", "B_Memory", "T_CD4", "TReg",
"T_CD8", "NK_T", "NK", "NK_CD56Hi",
"Monocyte_Classical", "Monocyte_NonClassical",
"Dendritic_Cells", "HSPCs", "Cycling_Cells"))
PBMC4[["Cell_Type"]] <- Idents(PBMC4)
UMAPPlot(PBMC4, cols = colors.use, label = F) + ggtitle("Cell Type Classifications")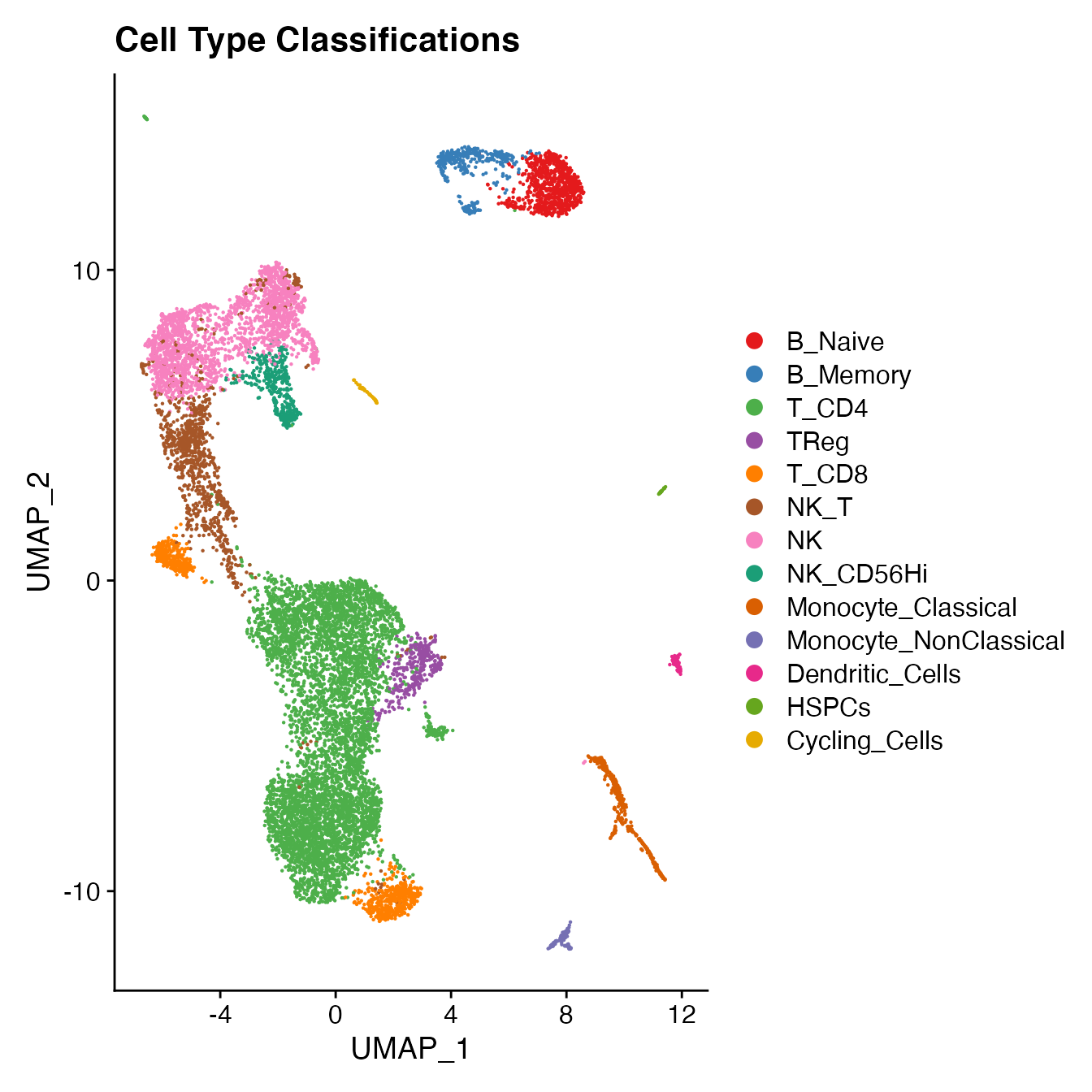
Compare single-sample workflow cell type classifications to joint classifications (generating a CMS)
# PBMC Sample 4-A
data(package = 'BatchNorm', PBMC4A_Single_ID)
S4Acms <- GetCMS(object = PBMC4,
sample.ID = "Sample_4A",
reference.ID = PBMC4A_Single_ID)
# PBMC Sample 4-B
data(package = 'BatchNorm', PBMC4B_Single_ID)
S4Bcms <- GetCMS(object = PBMC4,
sample.ID = "Sample_4B",
reference.ID = PBMC4B_Single_ID)
# Average CMS
mean(c(S4Acms, S4Bcms))## [1] 0.03208177Non-identical (duplicate, differing depths) Sequencing
Processing the Seurat Object
Data filtering & normalization
# Import unfiltered Seurat object (included with 'BatchNorm' package)
data(package = 'BatchNorm', PBMC5)
# Run "standard" Seurat workflow:
# Including filtering by mitochondrial percentage (+5 SD)
# Including data normalization, variable gene selection and gene scaling (performed on all samples together)
PBMC5 <- PBMC5 %>%
MitoFilter() %>%
NormalizeData(normalization.method = "LogNormalize", assay = "RNA", scale.factor = 10000) %>%
NormalizeData(verbose = FALSE, assay = "ADT", normalization.method = "CLR") %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData() %>%
RunPCA(npcs = 30)Selecting the appropriate number of Principal Components for UMAP reduction
# Identify correct numbers of PCs
## (Takes up to 5 minutes. Not run while rendering vignette for time)
# PBMC5.pca.test <- TestPCA(PBMC5)
# PBMC5.pca.test[, 1:20]
## 14 PCs with z > 1
## Proceed with 14 PCs for dimensional reduction & clustering
## Visualize PCs plotted by standard deviation:
ElbowPlot(PBMC5)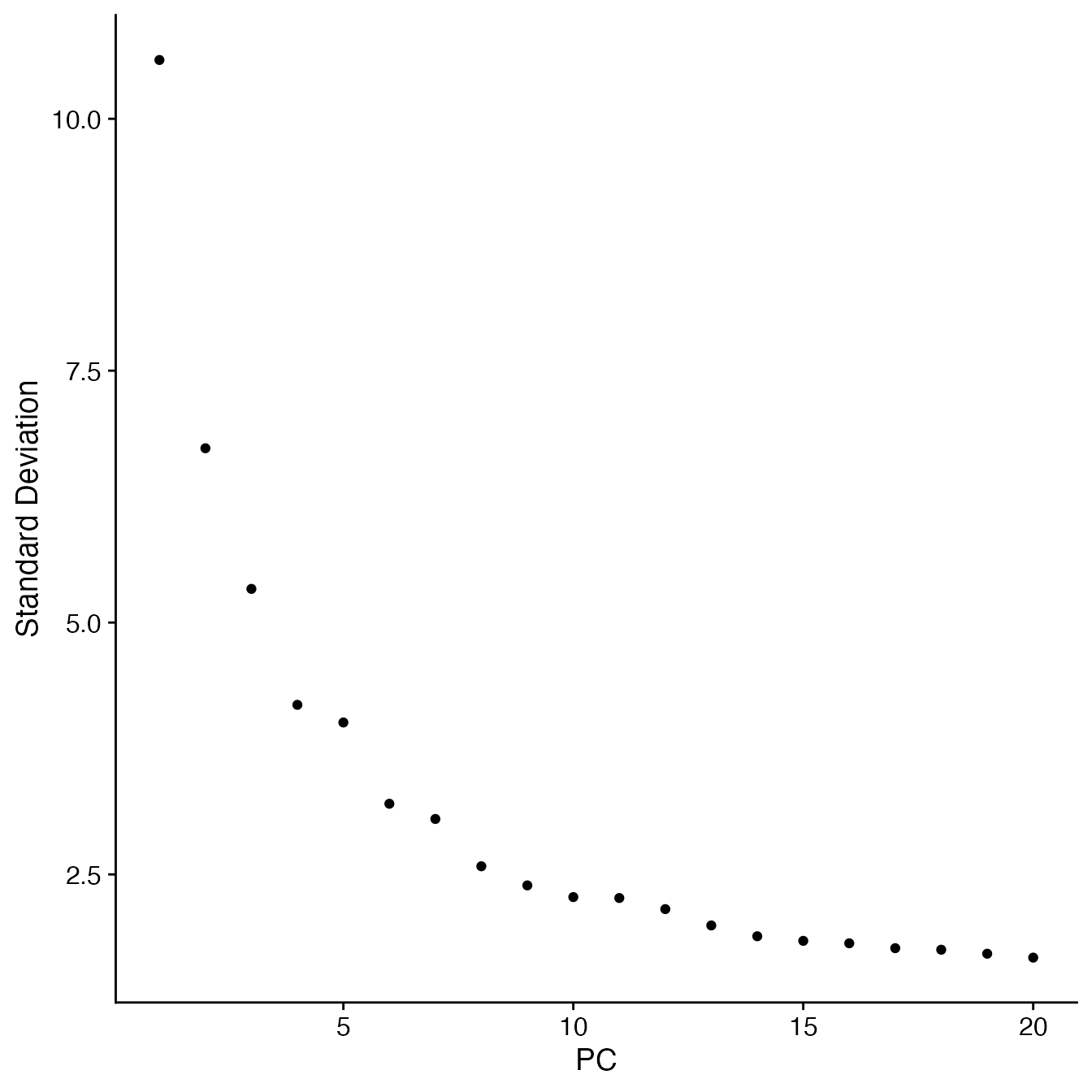
Generating a UMAP and clusters
PBMC5 <- PBMC5 %>%
RunUMAP(reduction = "pca", dims = 1:14) %>%
FindNeighbors(reduction = "pca", dims = 1:14) %>%
FindClusters(resolution = .8)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 7764
## Number of edges: 270395
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8881
## Number of communities: 19
## Elapsed time: 0 seconds
UMAPPlot(PBMC5, cols = colors.use, group.by = "orig.ident") + ggtitle("Default Workflow")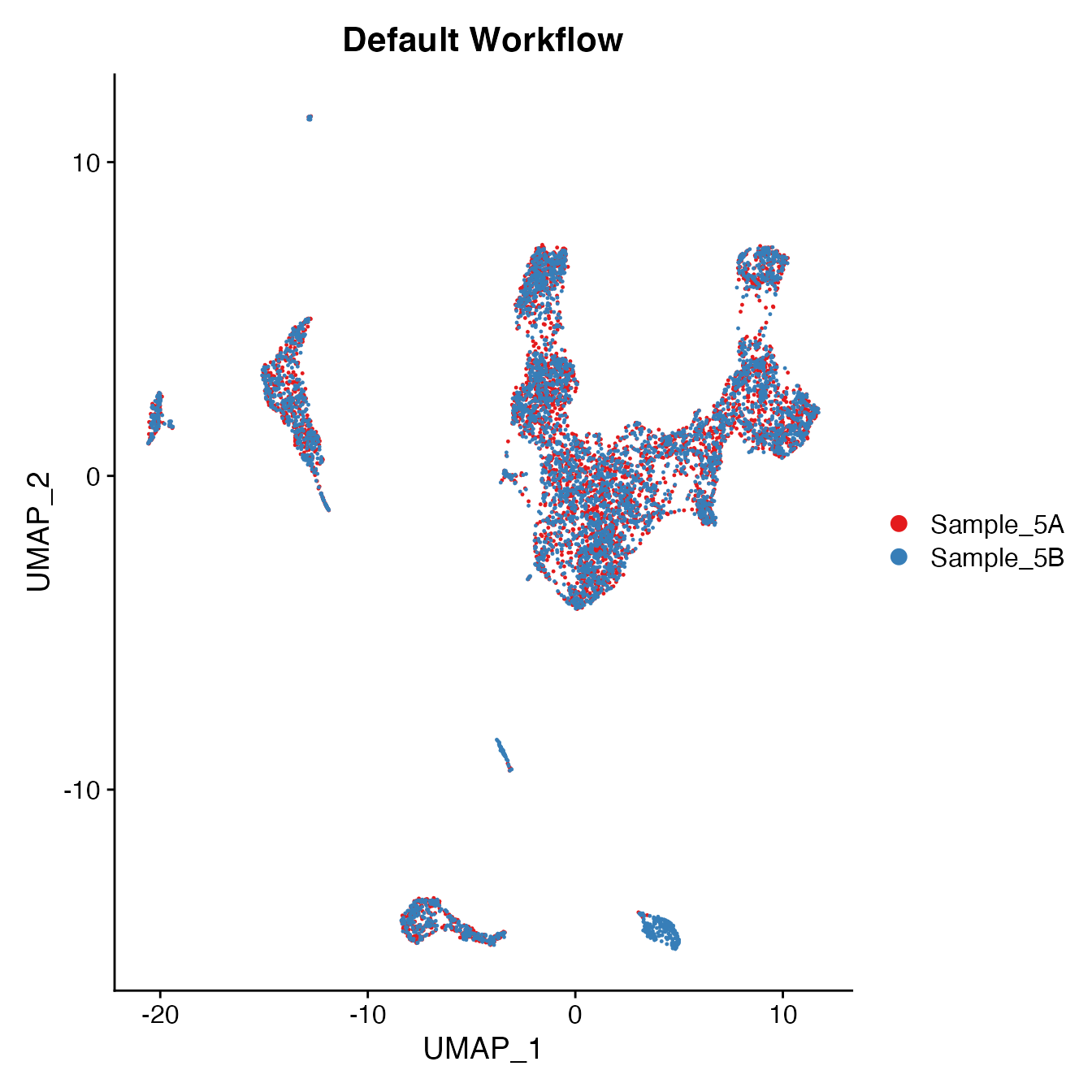
Measuring sample-UMAP integration (generating an iLISI score)
GetiLISI(object = PBMC5, nSamples = 2)## [1] 0.0275641Cell Typing of joint PBMCs object for CMS
Convert cluster classifications to cell type classifications
# For complete cell classification workflow see our vignette "Biaxial Gating of a Single Sample"
# More details can be found in figure S3 of our manuscript "Data Matrix Normalization and Merging Strategies Minimize Batch-specific Systemic Variation in scRNA-Seq Data."
UMAPPlot(PBMC5, cols = colors.use, pt.size = 2,
group.by = "seurat_clusters", label = T)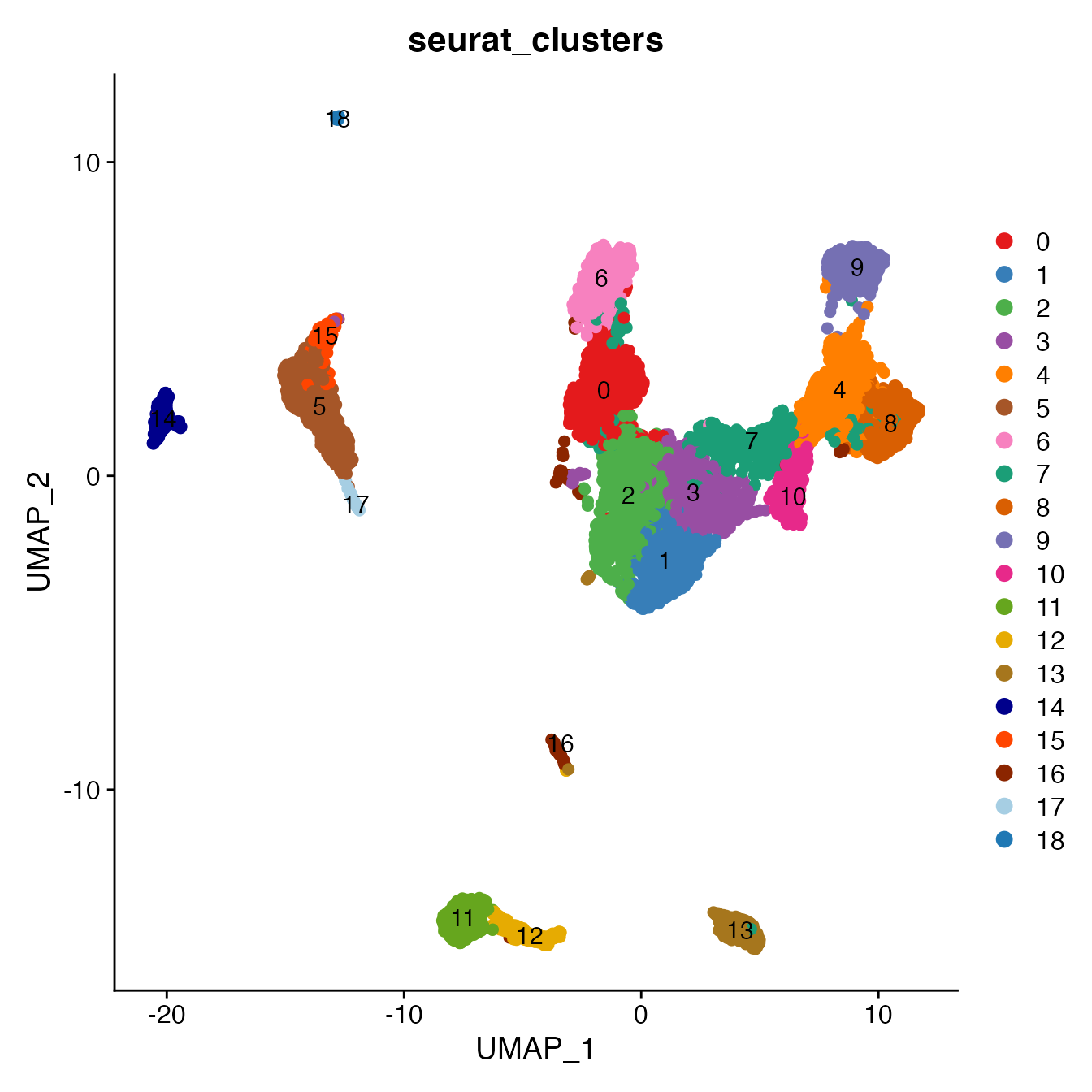
# B_Naive = 11
# B_Memory = 12
# T_CD4 = 0, 1, 2, 3, 16
# TReg not detected
# T_CD8 = 6
# NK_T = 4, 7, 8, 10
# NK = 9
# NK_CD56Hi = 9
# Monocyte_Classical = 5, 15
# Monocyte_NonClassical = 14
# Dendritic_Cells = 17, 18
# HSPCs = 13 (contains erythrocytes)
Idents(PBMC5) <- PBMC5[["seurat_clusters"]]
Idents(PBMC5) <- plyr::mapvalues(Idents(PBMC5), from = c(11, 12, 0, 1, 2, 3, 16,
6, 4, 7, 8, 10, 9,
5, 15, 14,
17, 18, 13),
to = c('B_Naive', 'B_Memory', 'T_CD4', 'T_CD4', 'T_CD4', 'T_CD4', 'T_CD4',
'T_CD8', 'NK_T', 'NK_T', 'NK_T', 'NK_T', 'NK',
'Monocyte_Classical', 'Monocyte_Classical', 'Monocyte_NonClassical',
'Dendritic_Cells', 'Dendritic_Cells', 'HSPCs'))
Idents(PBMC5) <- factor(Idents(PBMC5),
levels = c("B_Naive", "B_Memory", "T_CD4", "TReg",
"T_CD8", "NK_T", "NK", "NK_CD56Hi",
"Monocyte_Classical", "Monocyte_NonClassical",
"Dendritic_Cells", "HSPCs", "Cycling_Cells"))
PBMC5[["Cell_Type"]] <- Idents(PBMC5)
UMAPPlot(PBMC5, cols = colors.use, label = F) + ggtitle("Cell Type Classifications")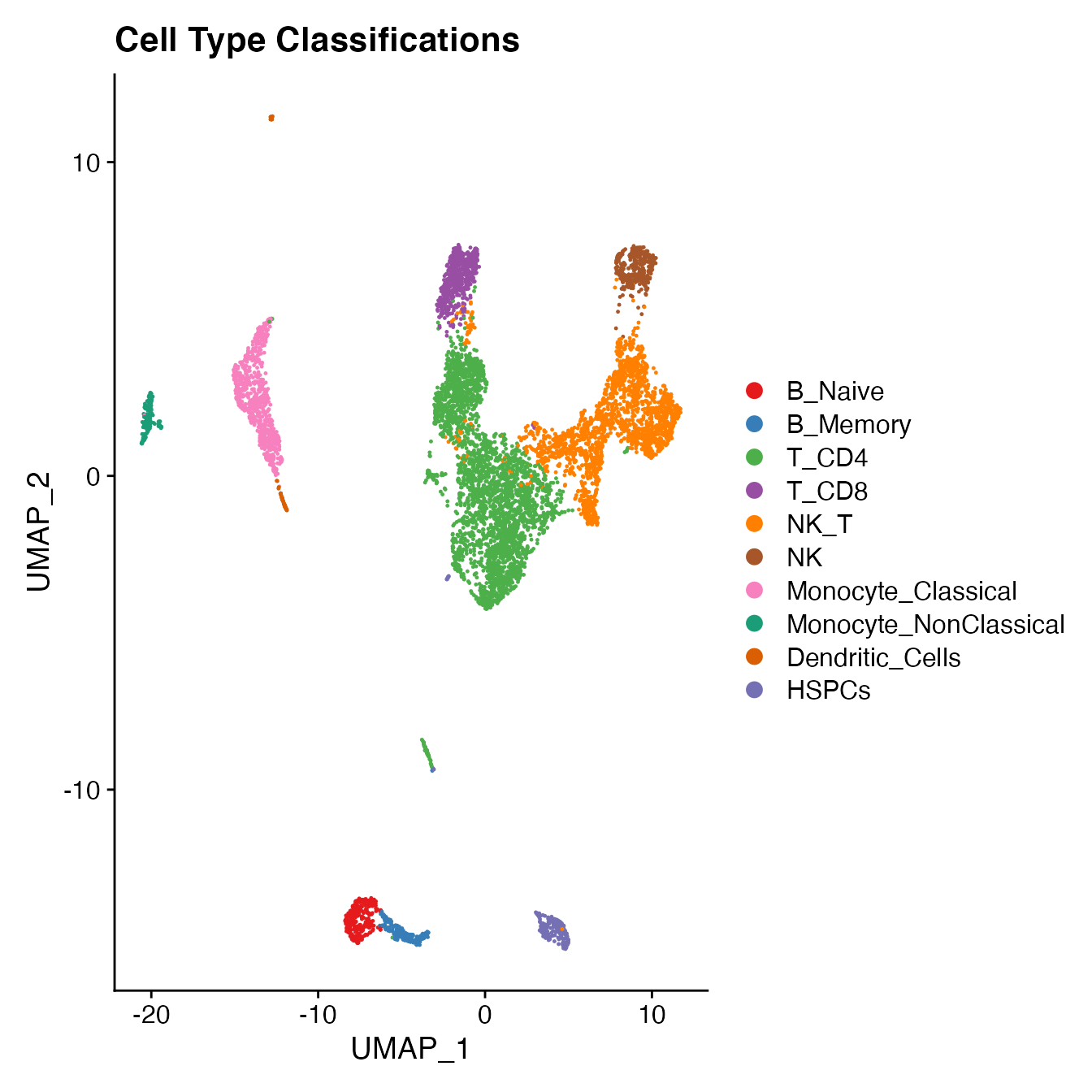
Compare single-sample workflow cell type classifications to joint classifications (generating a CMS)
# PBMC Sample 5-A
data(package = 'BatchNorm', PBMC5A_Single_ID)
S5Acms <- GetCMS(object = PBMC5,
sample.ID = "Sample_5A",
reference.ID = PBMC5A_Single_ID)
# PBMC Sample 5-B
data(package = 'BatchNorm', PBMC5B_Single_ID)
S5Bcms <- GetCMS(object = PBMC5,
sample.ID = "Sample_5B",
reference.ID = PBMC5B_Single_ID)
# Average CMS
mean(c(S5Acms, S5Bcms))## [1] 0.03288283